Затрагивающие небольшую часть населения патологии, такие как орфанные, относятся к заболеваниям, для лечения которых требуется поддержка со стороны правительства. Эти болезни связаны непосредственно с генетикой человека. Многие из них проявляются в детстве и сопровождают его в течение всей жизни.
Что такое орфанные заболевания?
Орфанные заболевания – это такие патологии, единого определения для которых в настоящий момент еще нет. Часть из них опирается на общее количество людей, которые на постоянной основе борются с основными симптомами. Другая часть определений учитывает доступность компенсационных возможностей для облегчения состояния больного человека.
В 1983 г. был принят Закон об орфанных препаратах. Его создание было предусмотрено с целью стимулирования исследовательских интересов широкого круга ученых. В конечном итоге он может стать причиной, из-за которой будут найдены или созданы способы полного восстановления больных людей.
В разных странах частота регистрируемых случаев для отдельных патологий довольно сильно отличается:
- Япония – 1 случай из 2500;
- США – 1 человек из 1500;
- Россия – 1 среди 10000.
В литературе принята общая статистика, которая считает отдельные типы заболеваний орфанными. По ее данным показатель находится в диапазоне от 1 из 1000 до 1 из 200000 человек.
Типы орфанных заболеваний
По закону в России такими патологиями изначально считались 86 патологий. Постепенно в их число были включены и другие. Количество орфанных заболеваний возросло до 215 в мае 2014 г.
В их число входят такие отклонения, с которыми абсолютное большинство жителей никогда не сталкивались в своей жизни, например, можно привести следующие болезни:
- саркомы костей, хрящевых и мягких тканей;
- раковые новообразования в глазах;
- онкология колоректальной зоны;
- различные типы лимфом;
- карцинома Меркеля;
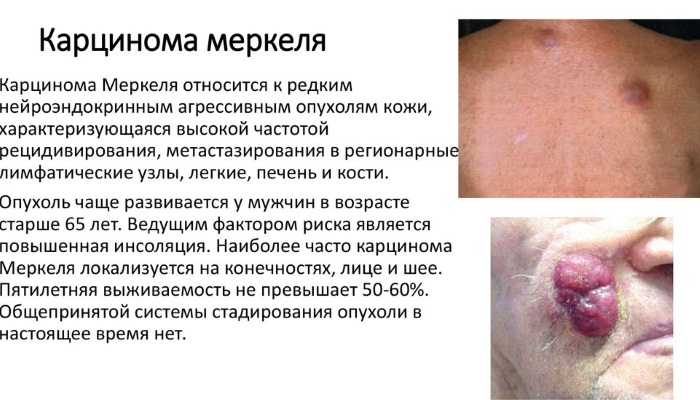
- миеломы;
- лейкозы.
Поскольку перечисленные заболевания в целом имеют похожую природу, но из-за имеющихся названий каждого конкретного типа все они вошли в число орфанных – редко регистрируемых патологий.
Причины
Орфанные заболевания – это ряд патологий, основная часть которых проявляется только в период взросления. В разных странах статистика значительно отличается. Во многом это связано с уровнем жизни местных жителей.
Например, в Индии количество больных людей значительно превышает аналогичный показатель в Европейских странах. Также на развитие болезней оказывает влияние генетика человека. Первые подозрительные симптомы могут быть обнаружены еще в младшем детском возрасте.
К причинам относятся следующие негативные факторы:
- высокий уровень радиационного фона;
- плохая экологическая обстановка;
- загрязнения окружающей среды;
- ослабленная иммунная система;
- наследственные факторы.
С учетом перечисленных причин бороться с риском их развития и улучшать статистику необходимо на высших уровнях со стороны правительства и основных действующих структур.
Причины у детей
Основной фактор – генетическая предрасположенность в сочетании с перенесенными вирусными инфекциями у матери в периоде вынашивания ребенка. Под влиянием этих причин нездоровый ген постепенно мутирует, после чего появляются первые симптомы – повод для серьезного обследования ребенка.
Перечень орфанных заболеваний
Орфанные заболевания – это опасные патологии, с которыми чаще всего врачи сталкиваются в детском возрасте пациента. Поэтому при появлении любых подозрений у часто болеющего ребенка можно предположить носительство генов, ответственных за развитие следующих болезней:
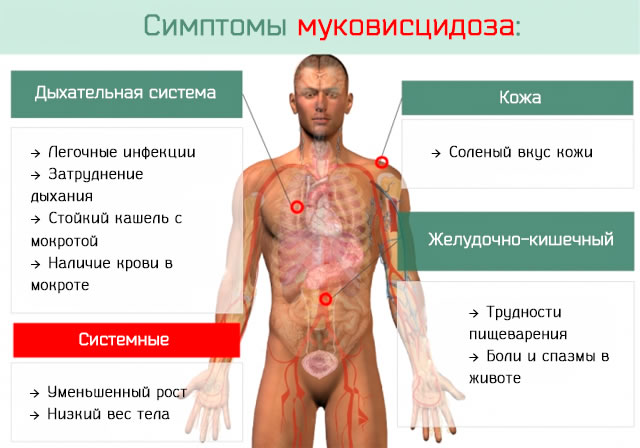
- муковисцидоз – самое распространенное системное отклонение, вызываемое мутацией специфического гена, трансмембранного регулятора муковисцидоза, результатом чего становится почти полная закупорка альвеол и дыхательная недостаточность, частота – 100:105;
- гемолитико-уремический синдром – при развитии заболевания появляются симптомы гемолитической анемии, тромбоцитопении и азотемии, к ним относятся диарея с кровью, боли в животе, бледная кожа, снижение количества мочи, отклонения в ЦНС и внутренних органах, частота – 1:105;
- пароксизмальная ночная гемоглобинурия – на фоне некоторых причин у больного начинают распадаться эритроциты в составе крови, при этом уровень гемоглобина увеличивается до такой степени, что организм запускает выведение избытка путем почечной фильтрации, частота – 0,55:105;
- апластическая анемия – при ее развитии угнетается функция кроветворения в красном костном мозге, в итоге наблюдается значительное снижение всех типов клеток в составе периферической крови, частота – 0,4:105;
- синдром Стюарта-Прауэра – наследственный недостаток фактора Х в плазме крови, белок под названием гамма-глобулин в норме содержится в ней на уровне 10 %, при падении ниже 1 % результатом становится летальный исход по причине утраты способности крови сворачиваться, частота – 0,2:105;
- тромбоцитопения пурпура – патология, сочетающая активное разрушение тромбоцитов до уровня менее 150х109 шт./л крови, в результате часто наблюдаются длительные и трудно останавливаемые кровотечения, частота – 24,6:105;
- дефект в системе комплемента – патология затрагивает комплекс белковых ферментов, которые требуются для нарушения мембран вредных микроорганизмов, поэтому симптомы схожи с недостатком иммуноглобулинов типа G, частота – 0,5:105;
- преждевременное половое созревание – при наличии такого состояния появление вторичных половых признаков, изменения скелета и другие проявления у девочек наблюдаются еще до 8 лет, у мальчиков – до 7 лет, частота – 4,0:105;
- фенилкетонурия – нарушения обмена ароматических аминокислот, фенилаланина и тирозина, клиническими симптомами которых становятся неврологические и психические расстройства вплоть до умственной отсталости и развитие микроцефалии в будущем, частота – 4,0:105;

- лейциноз – болезнь «кленового сиропа» относится к врожденным нарушениям обменных процессов, при которых в организме накапливаются 3 аминокислоты, лейцин, изолейцин и валин, в результате задерживается развитие, затем наступает фаза угнетения ЦНС вплоть до летаргии, частота – 15,6:105;
- нарушения обмена жирных кислот – при таком состоянии наблюдается дефицит синтеза карнитина, необходимого для переноса жирных кислот в митохондрии для дальнейшего окисления, симптоматически проявляется выраженной гипогликемией из-за резкого увеличения потребления глюкозы всеми тканями организма, частота – 2-10:105;
- гомоцистинурия – нарушения в системе обмена аминокислоты метионина приводят к поражениям нервной системы, зрительного аппарата, костей и суставов, множественному сосудистому тромбозу, частота – 0,4:105;
- тирозинемия – при этом заболевании накапливается недостаточность активности фумарилацетоацетат-гидролазы, в результате чего нарушается обмен тирозина, повреждаются ткани печени и почек, периферическая нервная система, отдаленно патология заканчивается циррозом печени и последующим летальным исходом, частота – 0,05:105;
- глютарикацидурия – в тканях организма происходит накопление глутаровой и 3-OH-глутаровой кислот, которые оказывают отравляющее действие на ЦНС, в частности, на структуры головного мозга под его корой, частота – 0,4:105;
- галактоземия – патология, при которой нарушаются обменные процессы в цепочке превращения галактозы в глюкозу из-за мутационных преобразований структурного гена, в итоге галактоза начинает разрушать ткани печени, нервной системы и хрусталик глаза, частота – 6,6:105;
- болезнь Фабри-Андерсона – отклонения в синтезе фермента альфа-галактозидазы А, который необходим для стабильной работы практически всех клеток, приводят к неизбежным, множественным повреждениям организма, частота – 1,75:105;
- болезнь Нимана-Пика – синдром, при наличии которого происходит накопление жира в различных органах, в частности, страдает печень, селезенка, головной мозг и лимфатические узлы с высоким риском летального исхода, частота – 0,85:105;
- различные ацидемии – метаболические расстройства, при которых наблюдается чрезмерное закисление крови, нарушение нормального обмена аминокислот, в результате которого происходит их накопление в организме, частота – 2-10:105;
- мукополисахаридоз – обменные сбои в процессе синтеза и утилизации гликозаминогиканов провоцируют развитие и накопление дефектов в костях, хрящах и соединительных тканях, частота – 0,6-1,3:105;
- печеночная порфирия – патология, при которой нарушается обмен пигментов в сочетании с увеличением концентрации порфиринов в крови и мягких тканях, может проявляться фотодерматитами, проблемами с кроветворением, расстройствами психики и функций ЖКТ, частота – 10,1:105;
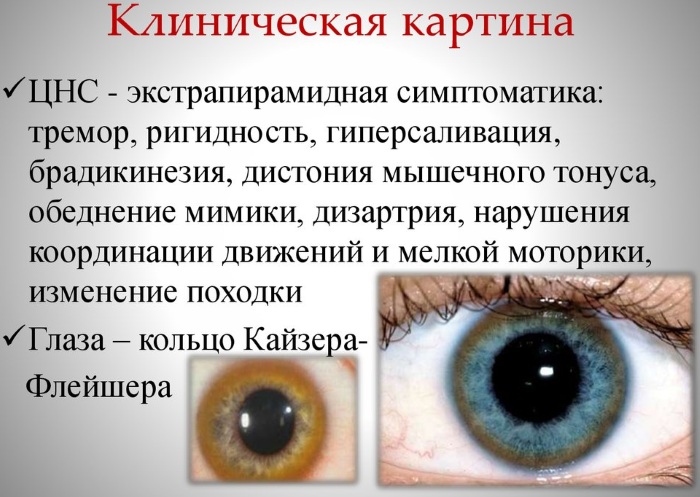
- болезнь Вильсона – неправильный обмен меди приводит к тяжелейшим поражениям периферической и центральной нервной системы и патологическим изменениям в структуре внутренних органов, циррозу печени, частота – 5,84:105;
- незавершенный остеогенез – при таком отклонении не происходит полноценного формирования костных тканей, что в последствии становится причиной повышенной хрупкости всех костей в организме, частота – 6,5:105;
- юношеский артрит с системным началом – ювенильный идиопатический артрит начинается с лихорадочного состояния в течение минимум 3-х суток, после чего к симптомам добавляются эритема, сыпь, генерализованное увеличение лимфатических узлов, плевриты и перитониты, частота – 4,2:105;
- первичная легочная гипертензия – заболевание сопровождается повышением кровяного давления внутри легочной артерии и усилением общего сопротивления в сосудах, среди симптомов наблюдается нехватка воздуха, тахикардия, нарушения сознания, боли за грудиной и другие проявления, частота – 0,4:105.
Орфанные заболевания, встречающиеся с приведенной частотой, являются усредненными данными. Год от года они меняются под влиянием генетических комбинаций при рождении детей и в результате естественной убыли населения. Это всегда влияет на статистику, которая собиралась ранее.
Орфанные заболевания и инвалидность
Подобные заболевания в ряде случаев становятся причиной для признания ребенка инвалидом. Однако в некоторых регионах и при наличии разной симптоматики у 2 детей или взрослых людей с одинаковым диагнозом результат будет полностью зависеть от решения медицинской комиссии.
Поэтому при наличии желания получить инвалидность потребуется выполнить несколько условий:
- собрать все необходимые справки и заключения;
- пройти всех рекомендованных специалистов;
- освидетельствовать результат в МСЭК.
По окончанию всех операций на руки будет выдана справка об инвалидности с указанием сроков ее действительности.
Некоторые люди и, особенно, родители маленьких детей волнуются о том, что статус инвалида помешает дальнейшей социализации. По факту подтвержденная группа нетрудоспособности гарантирует прохождение специальной реабилитационной программы и другие преимущества перед здоровыми людьми:
- подключение к кабельному телевидению на льготных условиях;
- дополнительные скидки на различные виды товаров и услуг;
- лояльные условия для поступления в учебные заведения;
- получение необходимых лекарственных средств;
- снижение тарифов на городскую телефонию;
- санатарно-курортный отдых и лечение;
- бесплатный городской транспорт;
- льготы на предоставление жилья;
- бесплатный доступ в интернет.
В случае ухода за ребенком-инвалидом родителям также предоставляются некоторые преимущества:
- пособие по уходу в размере 60 % от утвержденной минимальной оплаты труда;
- зачисление всего времени, проведенного с ребенком, в трудовой стаж.
Указаны основные критерии, которые остаются практически неизменными. Ряд других преимуществ периодически пересматривается правительством и местными властями, поэтому уточнять все детали рекомендуется непосредственно в региональных ведомствах.
Лечение
Орфанные заболевания – это такие патологии, для лечения которых предназначаются так называемые лекарства-сироты. Эти препараты используются для компенсации состояний, угрожающих непосредственно жизни больного человека или тогда, когда риск связан с приобретением инвалидности. В ряде стран число подобных больных не должно превышать установленного уровня.

Связано это с тем, что лекарства-сироты относятся к медицинским препаратам, которые экономически невыгодны ни производителю, ни государству. Но они в полной мере отвечают потребностям населения. По этой причине назначать лекарственное средство и выписывать все сопроводительные документы может только специализированный врач.
В России для лечения тяжелых патологий, несмотря на постоянный экономический кризис, выделяются крупные суммы финансовых средств для закупки медицинских препаратов. Поэтому при недостаточном доходе рекомендуется плотно заняться процедурой и предоставить в Медико-Социальную Экспертную Комиссию полный комплект подтверждающих документов.
Когда следует обратиться к врачу
Самое важное условие для максимально раннего выявления имеющихся нарушений, которые могут проявиться клиническими симптомами спустя несколько лет, — это пройти процедуру полного скрининга в период новорожденности. После этого рекомендуется регулярно посещать весь перечень врачей, которые будут рекомендоваться наблюдающим педиатром.
Это в разы увеличит шансы выявить патологию на доклинической стадии, а значит, и назначенное лечение станет в разы более эффективным. Также это позволяет избежать инвалидизации пациента и сохранить здоровье на уровне, достаточном для ведения полноценной повседневной жизни, включая годы обучения и дальнейшую трудовую деятельность по освоенной специальности.
Такой скрининг по действующим нормативам в России проводят в следующие сроки после появления ребенка на свет:
- у недоношенного – на 7 сутки;
- у доношенного – на 4 сутки.
Более ранний забор образцов крови может привести к получению ложноположительных результатов.
Скрининг позволяет выявить предрасположенность к развитию нескольких заболеваний:
- адреногенитальный синдром;
- врождённый гипотиреоз;
- фенилкетонурия;
- муковисцидоз;
- галактоземия.
Все они при несвоевременной диагностике и без подтверждения носительства соответствующего гена неизбежно вели к инвалидизации ребенка по мере его взросления. В ряде областей России список значительно расширен и может включать до 16 и более направлений. Например, в Москве с 2018 г скрининг проводится для выявления 11 заболеваний, включая перечисленные патологии.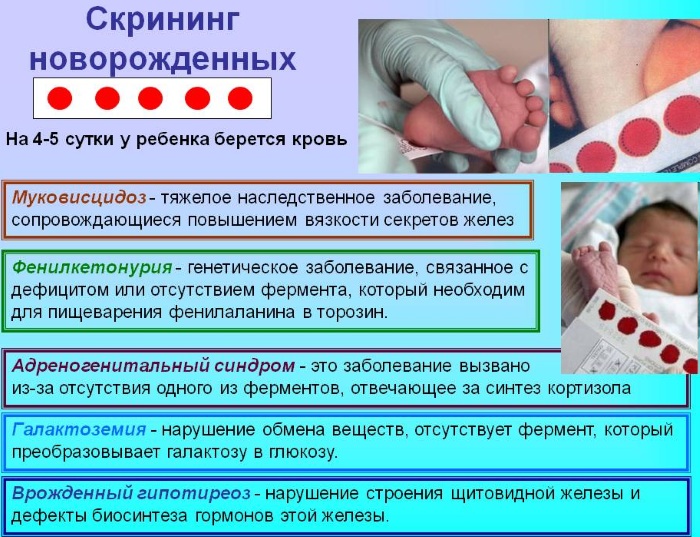
Процедура взятия образца крови не отличается сложностью для специалиста, но может сопровождаться немедленной реакцией ребенка в виде крика в ответ на внезапное ощущение боли.
Взятие крови на анализ именно из пятки обусловлено тем фактом, что пальцы на руках еще слишком малы, чтобы испытывать такие вмешательства. Мать должна спокойно подойти к выполнению необходимой процедуры, поскольку именно от нее во многом будет зависеть, как новорожденный перенесет неприятную манипуляцию.
В некоторых случаях женщины, далекие от медицины, не понимают, зачем делать скрининг, если никто из ближайших родственников не имеет никаких патологий. Необходимость связана с тем, что большинство самых тяжелых генетических заболеваний способны передаваться далеко не через 1 поколение. Поэтому о фактических рисках никто не сможет предоставить реальные данные.
Клинические рекомендации
Основной принцип терапии у больных с подтвержденными орфанными заболеваниями – патогенетическое лечение. Такой способ направлен на коррекционное восстановление функций поврежденных органов и устранение дефектов, вызванных конкретным геном:
- если ген не выполняет функций, за которые несет ответственность, их необходимо возместить;
- если ген производит ненужные эффекты с образованием ядовитых веществ, их избыток следует выводить.
На примере сахарного диабета выполнение такого принципа выглядит, как снижение уровня глюкозы в крови препаратами для внутреннего приема:
- толбутамид;
- карбутомид;
- глиформин;
- глюкофаж;
- адебит.
Другой вариант – использование препаратов с содержанием инсулина.
| Средства для патогенетического лечения | Типы используемых препаратов |
| Вещества для стабильной работы обмена веществ | Глюкоза, витаминно-минеральные смеси, гормональные таблетки, средства для стабилизации функций печени и препараты для нормального кислотно-щелочного и водно-электролитного баланса. |
| Вещества, оказывающие влияние на центральную нервную систему | Блокады на основе новокаина, барбитураты, нейролептики, успокоительные сборы или таблетки, средства для поднятия тонуса. |
| Детоксиканты и препараты для пищеварения | Гепатопротекторы, средства с содержанием ферментов и экстракта поджелудочной железы, ПНЖК и вещества, усиливающие детоксикационную способность печеночной ткани. |
| Вещества, стимулирующие образование и регулярное выведение мочи | Фуросемид, веро-спиронолактон, мочегонные травы, производные бензотиадизина, пиримидина и птеридина.
|

С иммуннозаместительной целью применяют следующие средства:
- неспецифические иммуноглобулины – 10 % смесь бета- и гаммаглобулинов кровяной сыворотки;
- селоколострин – вещество, выделяемое из молозива, полученного от коров в течение 1 суток после отела.
Иммуноглобулины используют для терапии врожденного иммунодефицита и недостаточного веса. Сероколострин содержит до 98 % этих же веществ, при лечении которыми эффективно стимулируется природная устойчивость организма человека к различным разрушающим воздействиям.
В плане генетического наследования, орфанные заболевания относятся к мало контролируемым типам. Это свидетельствует о том, что каждый человек, а в современном мире и каждый младенец, должен быть максимально обследован, начиная с периода новорожденности.
В таком случае при стабильном получении отрицательных результатов вероятность появления симптомов развития генетического заболевания, которое было включено в программу скрининга, будет стремиться к нулю.
Видео про орфанные заболевания
Что такое орфанные заболевания:
https://www.youtube.com/watch?v=5_abLuC4ub4