Синдром Арнольда-Киари – врожденный или (в более редких случаях) приобретенный порок развития спинно-головного мозга, а также костных структур в основании черепа.
В последние годы отмечается тенденция роста врожденных пороков развития, среди которых аномалии мозга достигают 30% в общей структуре заболеваемости. Практически всегда это нарушение сочетается с вторичными мозговыми изменениями (водянка, поражение желудочков и другие).
Что такое синдром Арнольда-Киари?
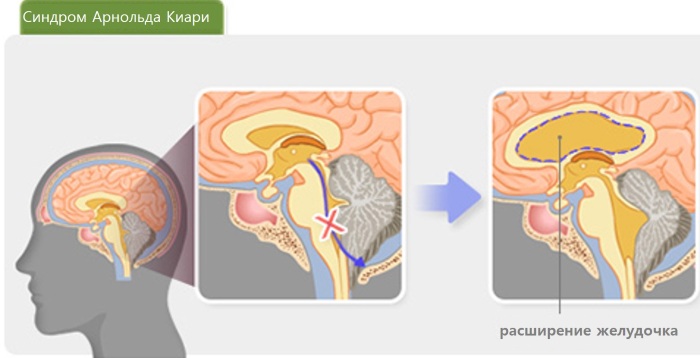
Синдром Арнольда-Киари – это тяжелая патология мозга, при которой выявляется опущение его ствола и миндалин мозжечка. В результате ухудшается циркуляция цереброспинальной жидкости, обеспечивающей постоянное внутричерепное давление, и водно-электролитного баланса.
Это способствует постепенному развитию гидроцефалии, внутричерепной гипертензии и возникновению кист в спинном мозге (до 80% пациентов).
Чаще всего данное нарушение формируется во внутриутробном периоде развития плода. Продолговатый мозг, регулирующий дыхание и кровообращение, сильно деформируется, а мозжечок, отвечающий за координацию и мышечный тонус, значительно отстает в своем развитии и постепенно наслаивается на продолговатый мозг.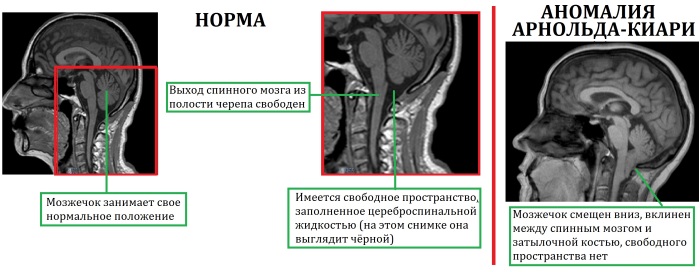
Сдавливание этих структур нередко приводит к двигательным и зрительным нарушениям, эпилептическим припадкам.
Эпидемиология
Распространенность данного нарушения достаточно высока – 1 новорожденный на 4-6 тысяч детей. Во взрослой популяции отмечается 3-8 случаев на 100 тыс. населения. Впервые эта аномалия была описана в медицине еще в конце XIX века после вскрытия умерших новорожденных.
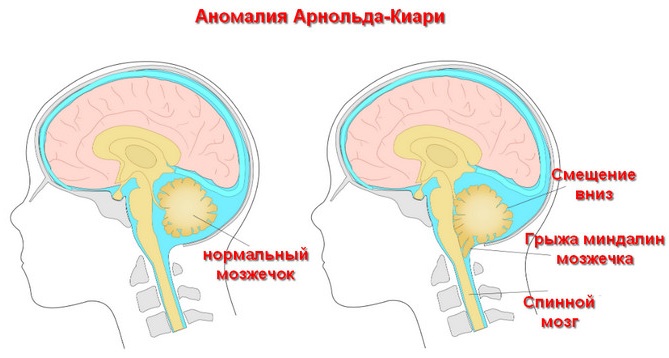
Так как у 95% пациентов мальформация Арнольда-Киари (от лат. malus – «плохой» и formatio – «формирование») сопровождается также образованием спинномозговой грыжи, то этот порок часто неправильно идентифицируется. Поэтому истинная распространенность может достигать больших значений. В 14-30% случаев заболевание является диагностической находкой во время медицинского обследования по другому поводу.
Типы и степени аномалии Арнольда-Киари
Синдром Арнольда-Киари – это заболевание, которое классифицируют на 4 типа:
- Легкий вариант течения болезни. Опущение мозжечковых миндалин в шейный отдел позвоночника ниже уровня большого затылочного отверстия, через которое полость черепа сообщается с позвоночным каналом. Выпячивание не превышает 12 мм. До 20% пациентов при этом страдают также гидроцефалией, а у половины появляются кисты в спинном мозге. Данный вид нарушения обычно проявляется впервые в юношеском возрасте, у более 50% новорожденных его течение бессимптомно. Чаще оно диагностируется у женщин, чем у мужчин.
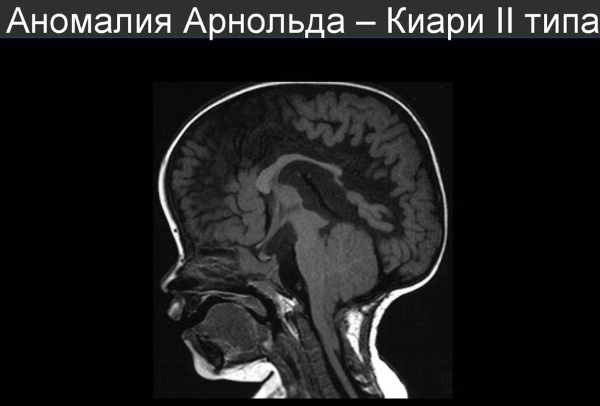
- Дислокация червеообразной структуры мозжечка, удлинение ствола продолговатого мозга и IV желудочка. Гидроцефалия при этом прогрессирует, сужается Сильвиев канал, соединяющий третий и четвертый желудочки, в спинномозговом канале возникает грыжа (95-100% пациентов). Выявляются также нарушения в развитии конечного мозга (включая большие полушария), промежуточного и среднего мозга, шейного отдела позвоночника; полное или частичное отсутствие мозолистого тела и заращения дужек позвонков в поясничном отделе. Этот вид отклонения диагностируется уже в период новорожденности. Отмечается высокая смертность во внутриутробном периоде и в раннем возрасте. Первые 2 типа болезни являются наиболее распространенными.
- Тотальная миграция структур заднего мозга в позвоночник, мозговая грыжа в затылке, выраженная внутричерепная гипертензия, увеличенный диаметр большого затылочного отверстия. У пациентов отмечается грубый порок развития черепа и головного мозга, при котором часть мозгового вещества находится вне черепной коробки. Также нередки сердечно-сосудистые патологии, заращение анального отверстия и другие отклонения ЖКТ и мочеполовой системы. Эта разновидность синдрома чаще всего не совместима с жизнью.
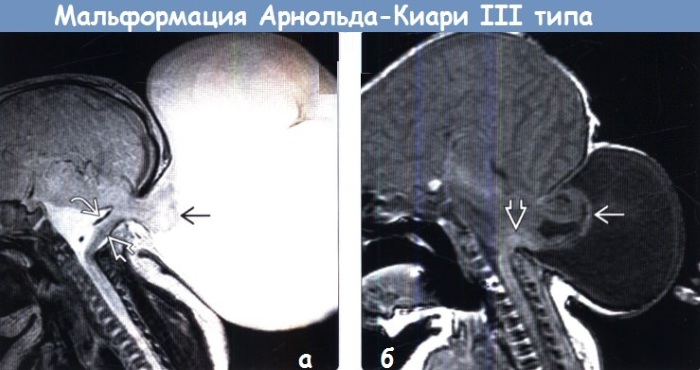
- Недоразвитие (гипоплазия) мозжечка без смещения его вниз вместе с продолговатым мозгом. Данное нарушение приводит к смерти новорожденного.
Некоторые специалисты классифицируют заболевание на 2 группы – с сирингомиелией (образованием полостей в спинном мозге) и без нее. Также выделяют переходную или пограничную стадию между I и II типом, которая сочетает симптомы обоих видов или представляет собой не полностью завершенный второй вариант развития болезни.
Причины и факторы риска у взрослых, детей
Механизм развития данной неврологической патологии связан со структурными изменениями в задней черепной ямке, в которой находится мозжечок, а также гидродинамическими нарушениями. Причины этого синдрома еще недостаточно изучены.
Врожденного характера
В основе врожденного дефекта лежит срастание задней стенки полости позвоночного столба с закладкой спинного мозга в основании черепа. При этом в процессе эмбрионального развития спинной мозг должен подтянуться вверх, но этого не происходит.
Продолговатый мозг и мозжечок «втягиваются» вниз, сквозь большое затылочное отверстие по направлению к позвоночному каналу. Иногда они обнаруживаются на уровне 4 шейного позвонка.
Факторами риска для развития аномалии у плода являются:
- поздний возраст беременной женщины;
- выкидыши в анамнезе;
- токсикоз во время беременности;
- плацентарная недостаточность, преэклампсия;
- контакт беременной женщины с вредными веществами;
- инфекционные заболевания в период вынашивания (включая болезни, передающиеся половым путем).
Однако, как показывают исследования, почти у половины женщин беременность протекает без осложнений.
Приобретенного характера
У взрослых опущение структур мозжечка может возникнуть из-за частых пункций в спинной мозг, которые проводятся в диагностических целях, а также при лечении некоторых заболеваний и в качестве спинальной анестезии. У части пациентов такое состояние формируется после имплантации шунтирующей системы для лечения гидроцефалии.
Симптоматика
Синдром Арнольда-Киари I типа может длительное время протекать бессимптомно.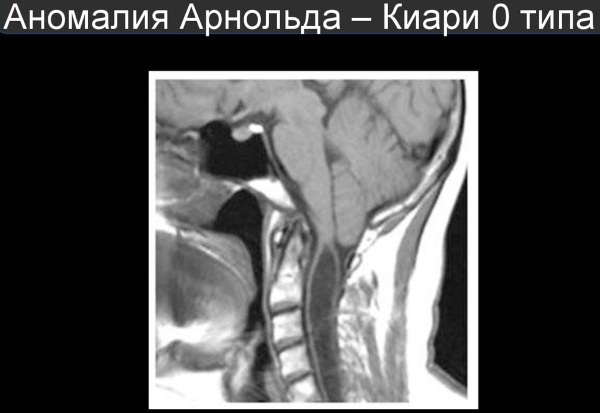
Это нарушение имеет следующие признаки:
- болезнь проявляется у подростков либо в 30-40-летнем возрасте;
- нарушения чувствительности, чаще всего в руках (40-76% пациентов);
- головная боль (наиболее распространенный симптом, отмечающийся у 47-73% больных);
- боль в шее и затылке, что усиливается из-за кашля, чихания, напряжения брюшных мышц;
- частые обмороки, падения, связанные с нарушением кровообращения спинного мозга, что вызывает резкое снижение мышечного тонуса; онемение конечностей;
- частое прекращение дыхания во сне более, чем на 10 секунд (50-70% пациентов), острая дыхательная недостаточность;
- общая слабость и снижение тонуса в конечностях;
- измененное вынуждено положение головы;
- скандирование речи (разделение на отдельные слова);
- колебательные движения глаз, преимущественно направленные вниз;
- нарушенная координация, походка, двигательная активность; рассогласованность, избыточность или недостаточность амплитуды движений;
- искривление позвоночника (50-75% пациентов);
- задержка роста и умственного развития у детей, иногда преждевременное половое созревание;
- приступообразный кашель (10-14% больных);
- нарушение мелкой моторики, дрожание конечностей;
- ухудшение слуха (одностороннее или двустороннее);
- врожденные анатомические особенности – короткая, «бычья» шея, воронкообразная грудь, асимметричная форма черепа, низкая граница оволосения на шее, деформация стоп, аномалии сосков, высокое стояние лопатки;
- раздвоение в глазах, разные размеры зрачков, слепые участки в поле зрения, опущение века;
- нарушение глотания, атрофия мышц языка, частое икание;
- редко – замедление сердечного ритма, острая сердечно-сосудистая недостаточность, гипогликемия.
Зависимо от степени поражения симптомы могут варьироваться от незначительных до ярко выраженных.
При мальформации II типа отмечаются следующие признаки:
- появление симптомов уже в период новорожденности;
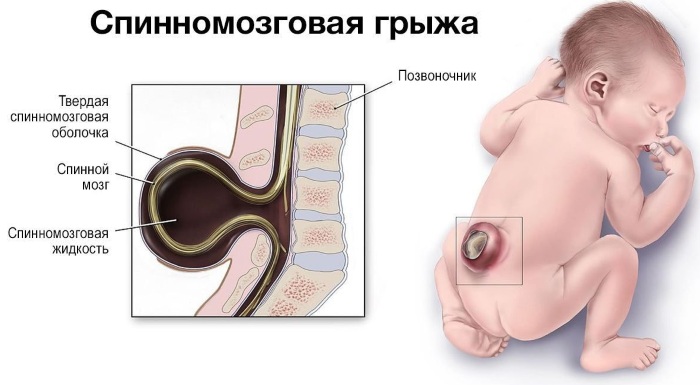
- спинномозговая грыжа;
- гидроцефалия (водянка головного мозга, увеличенная окружность головы, выбухающий родничок у новорожденных, косоглазие, высокая возбудимость, рвота, вялость, головные боли и головокружения);
- приступы удушья и остановки дыхания, в том числе во сне, свистящее шумное дыхание;
- ухудшенный глотательный рефлекс, затруднения при глотании (70% пациентов);
- слабость рук и мышц лица, снижение двигательной активности;
- судорожная поза с выгибанием спины;
- потеря голоса в результате пареза голосовых связок;
- дрожание глазных яблок, направленное вниз.
Для формы III варианта характерны такие симптомы, как:
- гидроцефалия;
- сердечно-сосудистые нарушения;
- патологии желудочно-кишечного тракта, мочевыводящей системы;
- головокружение;
- неустойчивость при передвижении;
- звон в ушах;
- головная боль;
- повышенный тонус в шейных мышцах.
Связанные расстройства
Данный синдром также часто сочетается со следующими патологиями:
- хроническое воспаление околоносовых пазух или киста в этой области;
- опухоли в мозжечке и мозолистом теле;
- киста турецкого седла;
- полное или частичное отсутствие мозолистого тела, соединяющего правое и левое полушария мозга;
- порок развития коры больших полушарий головного мозга, заключающийся в чрезмерно большом количестве мелких и частично сформированных извилин;
- нарушение формирования коры головного мозга (аномальное скопление серого вещества в различных областях, вызванное изменением миграции нейронов);
- пороки развития сердца и почек;
- недоразвитие базальных узлов, обеспечивающих двигательные и вегетативные функции;
- ранее закрытие черепных швов у детей, приводящее к ограничению объема черепа, его деформации и развитию внутричерепной гипертензии;
- полное или частичное сращением затылочной черепной кости и первого шейного позвонка (ассимиляция атланта);
- субатрофия (деструктивные процессы) полушарий головного мозга;
- смещение затылочной кости в сторону соединения верхнешейного отдела позвоночника с основанием черепа (базилярная импрессия);
- аномалия Робена, при которой выявляется врожденный порок челюстно-лицевой области (недоразвитие нижней челюсти, расщелина неба, западение языка);
- синдром Апера, при котором врожденная аномалия черепа сочетается со сращением пальцев рук и ног;
- синдром Вильямса, при котором изменяется строение лица, наблюдается умственная отсталость, а также другие генетические отклонения.
Осложнения
Синдром Арнольда-Киари – это патология, для которой характерна высокая смертность в раннем возрасте. Выраженность клинических признаков связана со степенью компрессии нервных тканей и ухудшением циркуляции спинномозговой жидкости.
У детей со II вариантом синдрома кроме смещения мозговых структур происходит сдавливание черепных нервов, что приводит к нарушению функции дыхания и глотания.
Увеличивается риск попадания пищи в легкие и развития аспирационной пневмонии, что и является причиной ранней смерти в большинстве случаев. Проведение хирургической операции помогает снизить неврологические расстройства и сохранить стабильное состояние не менее 2 лет у 94% пациентов.
Пороки III и IV типа являются самыми неблагоприятными, многие дети неоперабельны и умирают в первый месяц после рождения. У выживших отмечается паралич ног, нарушение работы тазовых органов, недержание мочи и кала, множественные пороки развития.
Диагностика
Для диагностики патологии используются следующие методы исследований:
- магнитно-резонансная томография головного мозга на стандартных режимах, при которой выявляются характерные аномалии, описанные ранее в статье;
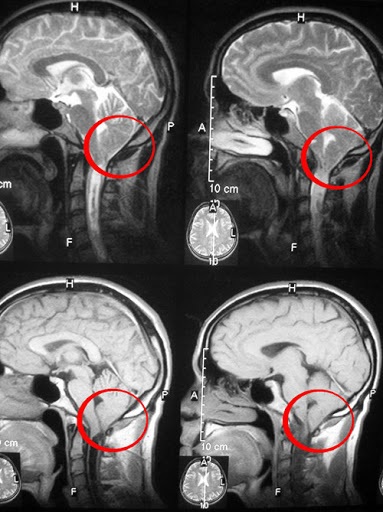
- фазоконтрастная магнитно-резонансная томография (ФКМРТ), позволяющая определить нарушения в движении ликвора – спинномозговой жидкости;
- нейросонография (ультразвуковое исследование мозга);
- краниография (рентгенологическое исследование костей черепа), наряду с предыдущим видом обследования она позволяет оценить степень уменьшения задней черепной ямки, увеличение большого затылочного отверстия и изменения в других внутримозговых структурах.
Внутриутробное обследование
Современная ультразвуковая диагностика может выявить эту патологию, начиная с 17-18 недели беременности. На ранних сроках точность определения нарушений составляет 24-45%. Чаще всего диагноз ставится во II-III триместрах.
Важно своевременно проходить скрининг, так как в этот период хорошо визуализируются полушария, мозжечок и участки расширения субарахноидального пространства, связанные с ним.
На УЗИ-снимках при этом визуализируются изменение формы и размеров мозжечка, его нечеткий контур, уменьшение полых образований между мозжечком и продолговатым мозгом.
Дополнительными диагностическими признаками также служат:
- гидроцефалия;
- увеличение желудочков головного мозга и изменение их формы (они становятся заостренными кзади);
- форма головы плода в виде «лимона» и мозжечка в виде «банана»;
- спинномозговая грыжа.
Сканирование должно проводиться в нескольких плоскостях.
Однако визуализация может затрудняться следующими факторами:
- ожирение у беременной;
- рубцовые изменения передней брюшной стенки;
- многоплодная беременность;
- неподходящее положение плода во время обследования;
- наличие других редких аномалий.
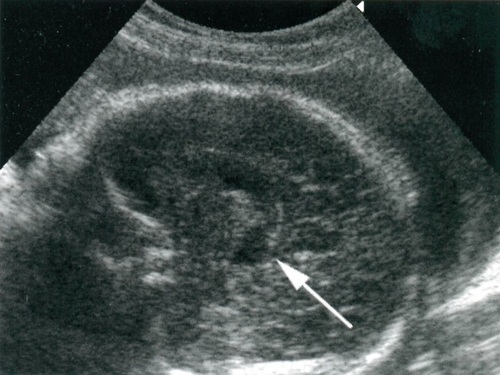 Для уточнения диагноза беременной женщине должно назначаться МРТ после 20 недели гестации. В первом триместре этот метод не используется, так как он может повлиять на процессы деления клеток эмбриона.
Для уточнения диагноза беременной женщине должно назначаться МРТ после 20 недели гестации. В первом триместре этот метод не используется, так как он может повлиять на процессы деления клеток эмбриона.
Лечение взрослых, детей при мальформации Арнольда-Киари
Тем пациентам, у которых выявлен синдром I типа без выраженных клинических симптомов, сохраняющихся без изменений на протяжении многих лет, показано динамическое наблюдение за состоянием здоровья. Контроль (включая рентгенологическое исследование, УЗИ или МРТ) проводится не реже 1 раза в год.
В остальных случаях, а также при ухудшении состояния больного показано хирургическое вмешательство.
Консервативная терапия
При незначительном болевом синдроме пациентам назначают симптоматические средства, описанные в таблице ниже.
| Название препарата | Основное фармакологическое действие | Суточная дозировка | Средняя цена, руб. |
| Глиатилин | Улучшение передачи нервных импульсов | По 400 мг 3 раза | 710 |
| Алпразолам | Противосудорожное, миорелаксирующее средство | По ¼ таблетке по 1 мг 3 раза | 850 |
| Флуоксетин | Антидепрессант | По 2 мг утром | 100 |
| Диакарб | Диуретик (мочегонное) | По 1 таблетке | 280 |
| Аспаркам | Улучшение метаболических процессов | По 1-2 таблетке 3 раза в день | 65 |
| Кетонал | Обезболивающее | 1-2 капсулы 2-3 раза | 170 |
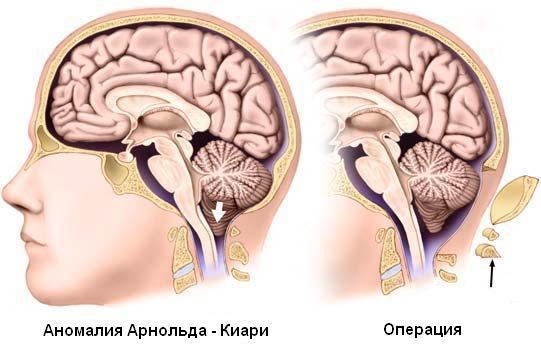
Хирургическое вмешательство
Синдром Арнольда-Киари – это патология, основным способом лечения которой является хирургическая операция. При наличии спинномозговой грыжи она проводится новорожденным в первые дни жизни.
Показаниями для операции являются:
- выраженная неврологическая симптоматика, значительно ухудшающая качество жизни и состояние пациента;
- прогрессирование сирингомиелии (образования полостей в спинном мозге, заполненных спинномозговой жидкостью);
- сильная головная боль из-за вклинивания и компрессии мозжечка.
Хирургическое вмешательство производится в следующем порядке:
- Предоперационная подготовка (показана для пациентов старше 60 лет, при обструктивных патологиях верхних дыхательных путей, острых и хронических сердечно-сосудистых заболеваниях, гипертонии, аритмии, гипертрофии левого желудочка сердца, других соматических патологиях). Содержание мероприятий зависит от вида заболевания и определяется индивидуально. Они направлены на снижение риска послеоперационных осложнений.
- Больного укладывают на живот, на бок или в положение полусидя, чтобы предоставить доступ к затылочной области. Сидячее положение показано пациентам с избыточной массой тела.
- Производится интубация (введение эндотрахеальной трубки в трахею) для обеспечения проходимости дыхательных путей и общая анестезия.
- Для обеспечения физиологического положения пациента используются валики, голова фиксируется как минимум в 3 точках.
- Осуществляется разрез кожи в шейно-затылочной области задним срединным доступом.
- Производится иссечение черепа, удаление части или всей дужки позвонка.
- При наличии патологических изменений арахноидальной оболочки головного мозга и спаек оболочки с мозжечком проводится вскрытие большой затылочной цистерны, иссечение спаек.
- При опущении мозжечка ниже позвонка С2 производится его резекция.
- При расширении четвертого желудочка осуществляется имплантирование шунтов.
- Выполняется пластика мозговой оболочки, для чего используется искусственный материал. В некоторых случаях применяется титановый имплант, чтобы устранить компрессию.
- Рана ушивается с помощью мышечного слоя.
Положительная динамика после операции наблюдается примерно у 70% пациентов. Она обратно пропорциональна продолжительности заболевания. У части прооперированных она отсутствует из-за необратимых изменений в нейронах. Чаще всего это наблюдается в тех случаях, когда от начала клинических проявлений прошло более 2 лет. 13-30% больным требуется повторная операция.
Осложнениями хирургического вмешательства могут быть:
- развитие инфекционных процессов;
- истечение цереброспинальной жидкости;
- плохое заживление раны;
- нестабильность шейного отдела позвоночника;
- образование гематомы (кровоизлияния), сдавливающей ствол мозга;
- опущение мозжечка из-за избыточной трепанации черепа;
- перелом полукольца позвонка С1.
Послеоперационная летальность обычно не превышает 2%.
Какие могут быть последствия?
Отсутствие своевременного лечения может привести к следующим последствиям:
- атрофия ткани мозжечка;
- разрушение мозговых нейронов;
- нарушение координации, двигательных функций;
- ухудшение зрения и слуха;
- паралич;
- у детей – нарушение нормального роста и умственного развития;
- вегетативные нарушения, приводящие к острой дыхательной и сердечной недостаточности;
- летальный исход.
Синдром (или мальформация) Арнольда-Киари вызывает тяжелые нарушения центральной нервной системы. Это заболевание в большинстве случаев носит наследственный характер, но может проявиться не сразу. К ранним методам диагностики относятся УЗИ и МРТ во время беременности.
Основной способ лечения – хирургический. В тех случаях, когда отсутствует выраженная симптоматика, предпочтительна выжидательная тактика и систематический контроль над состоянием пациента.
Видео о синдроме Арнольда-Киари
Аномалия Арнольда-Киари: